
 What is it HLH?
What is it HLH?
Hemophagocytic Lymphohistiocytosis (HLH) is a rare and often fatal syndrome of uncontrolled and ineffective inflammatory response to a certain trigger. It is characterized by excessive proliferation of lymphocytes and macrophages (histiocytes), hence the name “lymphohistiocytosis”. This results in the overproduction of cytokines, responsible for many of the clinical features present in this syndrome.
Familial, or genetic, HLH occurs as a result of a genetic mutation leading to impaired cytotoxic function. There have been several genetic mutations indicated in the development of HLH, including an association with congenital immunodeficiency syndromes, such as Chediak-Higashi, Griscelli and X-Linked Lymphoproliferative Syndromes. This form most often occurs within the first year of life (median age 8 months), with the majority of pediatric cases occurring <2 years of age, but can range from infancy to adulthood.
Acquired HLH occurs in the setting of an underlying condition, such as immunodeficiency, malignancy, or autoimmune disease. When HLH is secondary to a predisposing autoimmune disease, it is referred to as macrophage activating syndrome (MAS). Acquired HLH is the most common cause of this syndrome in adults, but this form can be seen in all ages. Overall, the syndrome is most often triggered by an infectious agent in an otherwise healthy person.
When to suspect HLH?
Clinically, acquired and genetic forms of HLH are often indistinguishable and one should have a high index of suspicion for any critically ill patient who presents with prolonged fever and unexplained cytopenia’s with multi-organ involvement.
HLH can often present like sepsis and often a full sepsis workup is appropriately initiated. Characteristics that may raise your suspicion in the emergency room or intensive care unit would be any critically ill patient with multiorgan involvement resembling infection or inflammatory disorder.
Often imaging may be obtained as part a separate workup such as CT scan or bedside ultrasound, and if signs such as splenomegaly, or hepatosplenomegaly are observed this should heighten your suspicion. Basic lab values such as a complete blood count (CBC) and liver function tests (LFT) may reveal cytopenia’s (including anemia, leukopenia or thrombocytopenia) as well as elevated liver enzymes. Elevated LFT’s are a common finding in adults and pediatrics, but not part of diagnostic criteria. Most of the imaging and labs are routinely ordered in the emergency department on initial presentation and can heighten your suspicion.
Additional patient history such as fever of unknown origin (FUO) and underlying autoimmune disease (ex. systemic juvenile idiopathic rheumatoid arthritis (formerly known as Stills Disease) should also help support your diagnosis.
If these above finding are present, consider adding additional tests such as a serum ferritin and triglyceride level. Both tests are easy to order and may help support your diagnosis of HLH sooner than later.
Table 1 lists the 8 diagnostic criteria for HLH which were derived from pediatric data, however both pediatric and adult HLH criteria remain the same. To make the diagnosis of HLH, one must have 5/8 criteria listed.
We will review each in detail, however 3 of the 8 tests are often not easy to order quickly and may take time. The soluble IL-2 receptor assay is usually a send-out lab and can take often up to a week to return, thus delaying the diagnosis. The NK-Cell activity is perhaps the most difficult to that measure, and many times not performed. A lymph node or bone marrow biopsy can sometimes be hard to obtain as it is an invasive procedure especially in the setting of a patient that may have high risk bleeding complications and critically ill.
Table 1: Diagnostic criteria: (Must fulfill 5 of 8 parameters)

Pathophysiology:
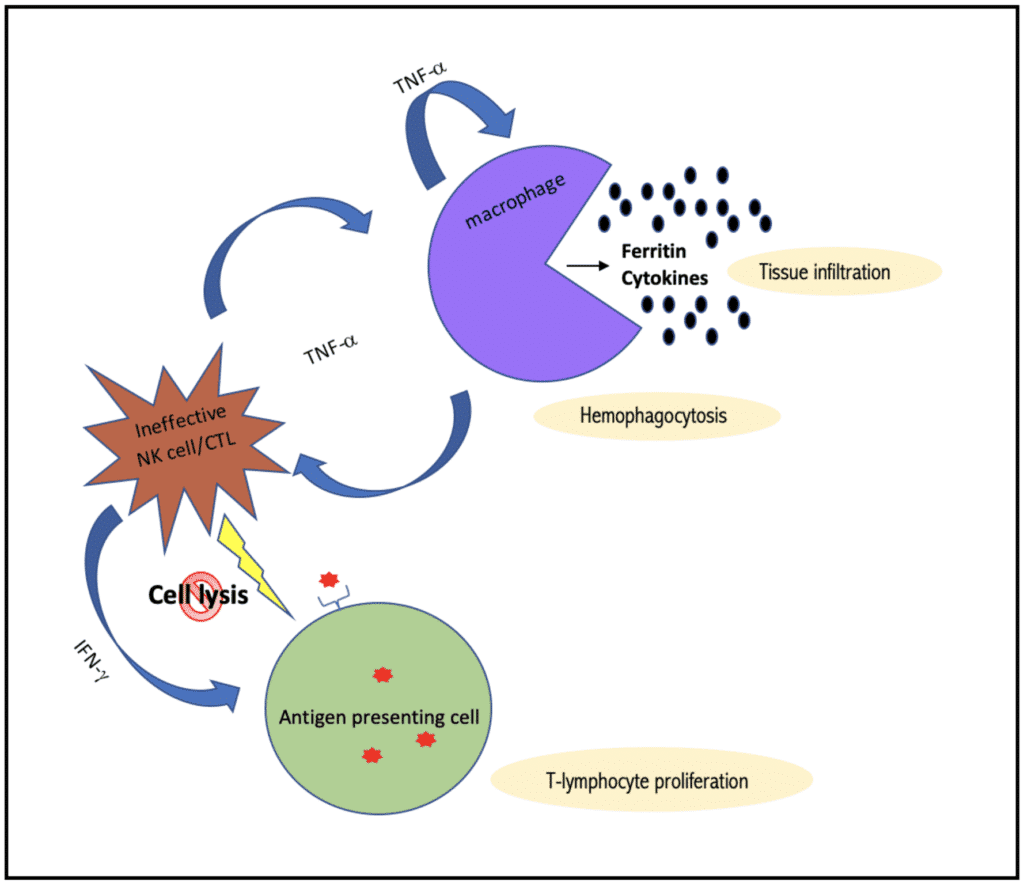
The pathophysiology of HLH can be simplified into three intertwined cellular processes:
- Genetic failure or transient impairment of cytotoxic cells (CD8-T-Cells and NK Cells) to induce cell lysis when encountered with an antigen presenting cell
- Hyperactivation of lymphocytes and macrophages due to the failure to terminate an immune response leads to host tissue destruction ( Ex. Macrophages get out of control and engulf host cells including RBC, Platelets, and WBC causing cytopenias thus the name hemo (blood) phagocytosis (eating).
- Excessive proliferation of inflammatory cytokines resulting in further immune stimulation and progressive multi-organ dysfunction
- Fever
Cytokines behave as endogenous pyrogens; specifically, IL-1, IL-6, and TNFa. Elevation of these cytokines affects the central thermoregulatory center (hypothalamus) and turns up the body’s thermostat via prostaglandin synthesis, resulting in fever.
- Splenomegaly
The spleen is the largest lymphoid organ of the body and therefore rich in immune cells. Organ infiltration by activated histiocytes and lymphocytes leads to splenomegaly, much like an enlarged, reactive lymph node.
- Hypertrigylceridemia
Cytokines IL-1, IL-11, TNFa, and IFNg inhibits lipoprotein lipase (LPL) and therefore suppresses triglyceride breakdown.
- Elevated Ferritin
The production of a cytokine called growth differentiation factor 15 (GDF-15) leads to an increased availability of the key iron transporter, ferroportin, by downregulation of inhibitory protein, hepcidin. Upregulation of ferroportin leads to increased iron export out of cells, causing hyperferritinemia.
Elevated Ferritin levels are more sensitive and specific in pediatrics compared to adults. Allen et al found that ferritin levels >10,000 µg/L was 90% sensitive and 96% specific in the diagnosis of HLH. Whereas with adults, elevated Ferritin levels are less specific for the diagnosis of HLH. It’s important to note that the diagnostic criteria were derived from pediatric data. Schram et al demonstrated that no level in adults was specific for HLH and even with levels > 50,000 µg/L, the diagnosis of HLH was made in <20% of patients.
- Low Natural Killer (NK) cell activity
One of the most important factors in the pathogenesis of HLH is low or absent NK cell activity. This can often be one of the more difficult tests to obtain and for that reason is seldomly ordered. NK cells play a critical role in our innate immunity. They have an unsurpassed ability to kill infected cells because they do not require MHC for antigen presentation, as other lymphoid cells do. Typically, NK cells are recruited to fight by cytokines released from macrophages. However, in HLH, there is either genetic loss or transient dysfunction of the cytotoxic role of NK cells.
- Hemophagocytosis
Remember that histiocytes are simply tissue-bound macrophages with the same phagocytic capacity. Activated and indiscriminant histiocytes within the bone marrow and other tissues results in hemophagocytosis, leading to decreased number of erythrocytes, thrombocytes, and their precursors.
- Cytopenias
There are thought to be multiple causes of cytopenias in HLH. First, a somewhat intuitive cause is bone marrow infiltration and hemophagocytosis of myeloid precursors.
Additionally, cytokines such as TNFa may directly bind to receptors on the surface of stem cells and suppress hematopoiesis by apoptosis or quiescence. Finally, elevated ferritin in an inflammatory state may have a suppressive effect on erythroid progenitor cells.
Elevated soluble IL2 receptor (CD 25)
Soluble IL2 receptor (CD 25) is transmembrane protein present on activated T-cells and gets cleavage by activated macrophages. Therefore, elevated soluble IL2 receptor (CD 25) serves as a serum marker, becoming elevated in a lymphoproliferative state such as HLH.
Management of HLH?
The focus of this article is to discuss; (1)the subtypes of HLH (familial vs acquired), (2) the underlying pathophysiology, and (3) the diagnostic criteria and differences between the presentation in adults and pediatrics. Armed with this information, the hope is this may assist you in making a prompt diagnosis and start therapy early in a patient’s course.
HLH is considered a rare disorder but may be more common than we think as it may be often under recognized. Once your clinical suspicion is high, urgent consultation with Hematology-Oncology is critical to deliver optimal therapy. Familial HLH is thought to be universally fatal without therapy and acquired HLH also carries a high mortality without early treatment. Below is an overview of management strategies:
- Supportive care
- Directed towards organ failure, coagulation defects, anemia, thrombocytopenia, and treatment of opportunistic infections.
-
Trigger-directed treatment. The most common triggering conditions include:
- Infection, especially EBV, but other viral triggers as well including CMV, Influenza. Appropriate empiric therapy should be initiated promptly based on suspected organism. Rituximab is initiated for confirmed or probable EBV.
- Rheumatologic conditions, including JIA and SLE.
- Hematologic malignancy. HLH-specific therapy should be initiated along with malignancy-directed chemotherapy.
-
Controlling immune dysregulation
- Anti-IFNg monoclonal antibody, Emapalumab, is an FDA-approved therapy for relapsing or refractory HLH in both pediatric and adult populations.
-
HLH-specific therapy
- HLH- directed therapy includes etoposide, intrathecal methotrexate, and corticosteroids. This is protocol-based and warrants Hematology consult.
-
Hematopoietic stem cell transplant (HSCT)
- In inherited forms of HLH, HSCT is often necessary for a long-term cure and prevention of recurrence.
- HSCT is also indicated for patients with relapse or lack of response to therapy, malignancy-induced HLH, or CNS involvement.
Guest Post By:

References:
- Brusko TM et al. Influence of membrane CD25 stability on T lymphocyte activity: implications for immunoregulation. PLoS One. 2009. PMID: 19956753
- Janka GE et al. Hemophagocytic syndromes–an update. Blood Rev. 2014. PMID: 24792320
- Jiang F et al. Regulation of hepcidin through GDF-15 in cancer-related anemia. Clin Chim Acta. 2014. PMID: 24384540
- Kernan KF et al. Hyperferritinemia and inflammation. Int Immunol. 2017. PMID: 28541437
- Allen CE et al. Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis. Pediatric Blood Cancer 2008 PMID: 18085676
- Schram AM et al. How I treat hemophagocytic lymphohistiocytosis in the adult patient. Blood. 2015. PMID: 25758828
- Kmieciak M et al. Human T cells express CD25 and Foxp3 upon activation and exhibit effector/memory phenotypes without any regulatory/suppressor function. J Transl Med. 2009. PMID: 19849846
- Morikawa K et al. A role for ferritin in hematopoiesis and the immune system. Leuk Lymphoma. 1995. PMID: 8528049
- Morimoto, A et al. Hemophagocytic lymphohistiocytosis: Pathogenesis, diagnosis, and management. Pediatrics International. 2016 PMID: 27289085
- Querfeld U et al. Effects of cytokines on the production of lipoprotein lipase in cultured human macrophages. J Lipid Res. PMID: 23378601
- Wang W et al. Serum ferritin: Past, present and future. Biochim Biophys Acta. 2010 PMID: 20304033
- Yang, C., Zhu, X., Zhang, T et al. EBV-HLH children with reductions in CD4+ T cells and excessive activation of CD8+ T cells. Pediatr Res 2017. PMID: 28738028
Post Peer Reviewed By: Salim R. Rezaie, MD (Twitter: @srrezaie)
The post Hemophagocytic Lymphohistiocytosis (HLH): A Zebra Diagnosis We Should All Know appeared first on REBEL EM - Emergency Medicine Blog.