
 Today, I gave a lecture on Hemophilia to our residents in San Antonio, TX. Now this was a core content lecture that I have actually never given before. As I was preparing the lecture I realized that this is a diagnosis that comes up frequently enough that it is important to know about, but also so infrequently that I always have to look up the factor replacement options and calculations. So instead of being our typical evidence based evaluation of literature, this post will serve as a reminder of the basics of hemophilia and what are the essential elements one needs to know to appropriately treat a patient with Hemophilia.
Today, I gave a lecture on Hemophilia to our residents in San Antonio, TX. Now this was a core content lecture that I have actually never given before. As I was preparing the lecture I realized that this is a diagnosis that comes up frequently enough that it is important to know about, but also so infrequently that I always have to look up the factor replacement options and calculations. So instead of being our typical evidence based evaluation of literature, this post will serve as a reminder of the basics of hemophilia and what are the essential elements one needs to know to appropriately treat a patient with Hemophilia.
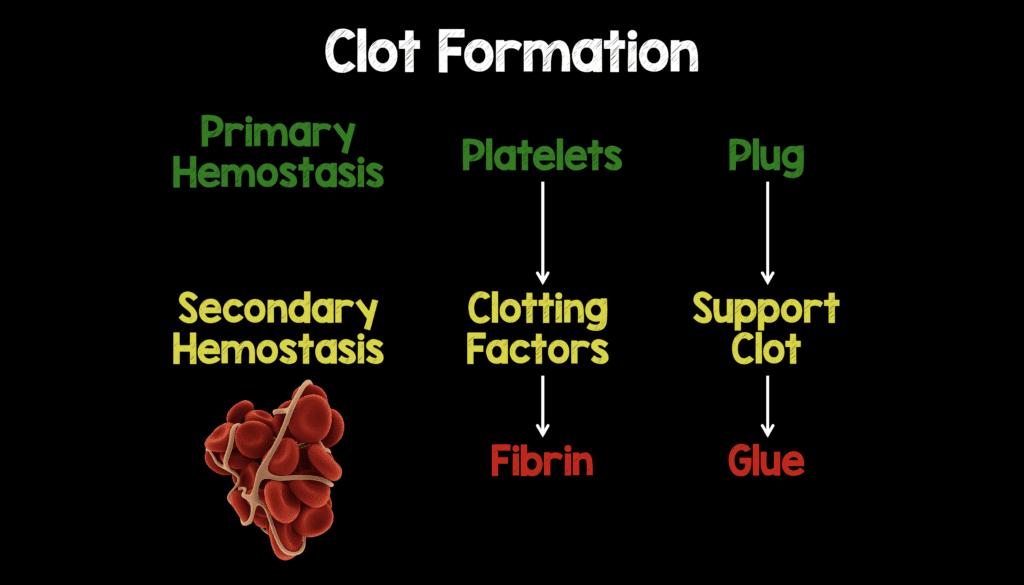
Lets Start with Some Basics of Clot Formation
Clot formation can essentially be broken down to endothelial injury, primary hemostasis (platelet function), and secondary hemostasis (clotting factors/coagulation cascade).
- When the vascular lining is injured, endothelial cells release von Willebrand factor (vWF) which allows platelet binding.
- Primary Hemostasis –> Platelet aggregation forms an initial plug (immediate) –> Deficiencies tend to cause superficial bleeds (i.e petechiae, ecchymosis, & purpura)
- Secondary Hemostasis –> Clotting Factors help support the plug (delayed) –> Deficiencies tend to cause deep bleeds (i.e hemarthroses, deep muscle bleeding)
- Fibrin is the last step in clot formation that acts as the glue to keep the clot together
Hemophilia typically has a prolonged aPTT but normal PT
- Hemophilia A (Factor VIII deficiency) and Hemophilia B (Factor IX deficiency) occur in the intrinsic pathway (aPTT) while Factor VII occurs in the extrinsic pathway (PT).
- Bleeding time and platelet counts are typically normal in Hemophilia A and B

Background and Clinical Presentation
Hemophilia is a set of X-linked recessive disorders of clotting factors. Deficiencies in factor VIII and factor IX produce hemophilia A and B respectively. The clinical manifestations of hemophilia A and B are practically indistinguishable and characterized primarily by easy bruising and recurrent bleeding into joints and deep muscles. Hemarthrosis is the most common complication of hemophilia with elbows, knees, and ankles most often affected. An important point to emphasize is patients may develop pain in the affected joint before any clinical signs can be detected, so it is important that even vague joint symptoms be taken seriously.
Intracranial hemorrhage (ICH) is the most common noninfectious cause of death in hemophiliacs. A history of trauma may be part of the presentation but severe hemophiliacs are at high risk for atraumatic ICH. Any patient with known hemophilia presenting with a history of or suspected head trauma or signs/symptoms suspicious for ICH should be assumed to be bleeding and managed aggressively with consideration of emergent factor replacement and possible admission for observation and neuro checks.
Hemophilia A (Classic Hemophilia)
- Deficiency in Factor VIII
- ≈80% of Hemophilia Cases (Factor VIII has 186,000 base pairs and therefore more prone to mutations than Factor IX)
- 1 in 5,000 male births
Hemophilia B (Christmas Disease)
- Deficiency in Factor IX
- ≈20% of Hemophilia Cases (Factor IX has 34,000 base pairs and therefore less prone to mutations than Factor VIII)
- 1 in 30,000 male births
Acquired Hemophilia
- These are patients with inhibitors to clotting factors making treatment more difficult.
- Causes:
- Malignancies (CLL, Adenocarcinomas)
- Pregnancy or Postpartum State
- Autoimmune Disorders (SLE, RA)
- Idiopathic
- Clotting Factor Mixing Test
- Mix normal plasma with patients serum
- Normal = aPTT will normalize
- Inhibitor = aPTT remains prolonged
- Low titers of inhibitor (≤5 Bethesda Units) can still be treated with Factor VIII concentrates
- High titers of inhibitor (>5 Bethesda Units) may require treatment with factor VIIa concentrates or prothrombin complexes (PCCs)
Factor Replacement
- The mainstay of treatment for hemophilia remains replacement of deficient clotting factors.
- Essentially 2 Types of Factor
- Recombinant Derived: Genetically engineered factor
- Plasma Derived: Cheaper than Recombinant, but Theoretical Infection Risk (i.e. HIV)
- Primary Prophylaxis vs Secondary Prophylaxis
- Primary Prophylaxis: Scheduled dosing of factor replacement for patients with severe disease to reduce the likelihood of spontaneous bleeding (What patients do as outpatients)
- Secondary Prophylaxis: Dosing of factor replacement based on acute need (i.e. scheduled surgical procedure, or in response to an injury)
- 1st ask the patient what kind of factor they are on and if they have their factor with them (Always try and use what the patient is on as a first choice)
- Factor VIII
- Treatment of choice for moderate to severe disease in Hemophilia A
- Factor VIII = Each U/kg = an Increase of Factor by 2%
- Factor IX
- Treatment of choice for moderate to severe disease in Hemophilia B
- Factor IX = Each U/kg = an Increase of Factor by 1%
- Factor VIIa
- Indicated for patients with high titer inhibitor levels
- Cryoprecipitate
- Not recommended for routine use in the US by the National Hemophilia Foundation (Due to high levels of viral transmission)
- 80 – 100 U of factor VIII per single donor bag
- Used if factor VIII concentrates are not available
- DDAVP
- Synthetic analog of vasopressin
- Drug of choice for acute bleeding or prophylaxis in mild hemophilia A
- Can increase factor VIII by 3x – 5x (Onset 30 minutes; peak 90 – 100 minutes)
- Dose: 0.3 mcg/kg/dose IV
- Fresh Frozen Plasma (FFP)
- 1 U of factor VIII per mL of FFP (1U FFP raises the factor level by only 3 – 5%)
- Volume overload is a limiting factor
- Prothrombin Complex Concentrates (PCCs)
- Contain factors II, VII, IX, and X
- Potential for thrombogenic side effects
Treatment Calculations
- Severity of Disease is Classified by Factor Levels
- Normal = 50 – 150% Factor Activity (0.5 – 1.5 IU/mL)
- Mild (60% of Cases) = 5 – 40% Factor Activity (0.05 – 0.4 IU/mL)
- Moderate (15% of Cases) = 1 – 5% Factor Activity (0.01 – 0.05 IU/mL)
- Severe (25% of Cases) = <1% Factor Activity (<0.01 IU/mL)
- In reality as EM Physicians what we need to know for treatment is the patients Factor Level:
- Option A: The patient knows their factor level
- Option B: Patient Does Not Know their factor level (If the patient does not know their factor level assume 0%)
- Severity of Bleed
- Mild to Moderate –> Soft Tissue, Muscle, Hemarthrosis, Epistaxis –> Factor Replacement up to 50% (It is actually more complex than this, but for sake of simplicity this will work)
- Severe –> CNS, GI, Neck/Throat, Major Trauma (i.e. Life Threatening Bleeds) –> Factor Replacement up to 100%
-
Putting it all Together (Calculations to Replace Deficient Factor)
- Units of Factor VIII Required = Patients Weight (kg) x 0.5 x (% Activity Desired – % Intrinsic Activity)
- Units of Factor IX Required = Patients Weight (kg) x 1.0 x (% Activity Desired – % Intrinsic Activity)
Clinical Take Home Points
- Deficiencies in clotting factors (secondary hemostasis) tend to cause deep bleeds (i.e hemarthroses, deep muscle) vs superficial bleeds (petechiae, purpura)
- Hemophilia typically has a prolonged aPTT (Intrinsic pathway – Factors VIII & IX) but normal PT (extrinsic pathway – Factor VII)
- The mainstay of treatment for hemophilia remains replacement of deficient clotting factors (Always try and use what the patient is on as a first choice)
- If a patient does not know their factor level, assume it is 0%
- Mild to Moderate bleeds should have their factor replaced up to 50%
- Life threatening/severe bleeds should have their factor replaced up to 100%
References:
- Bhat R and Cabey W. Evaluation and Management of Congenital Bleeding Disorders. Emerg Med Clin North Am. 2014; 32 (3): 673 – 90. PMID: 25060256
- Sadowitz PD et al. Hematologic Emergencies in the Pediatric Emergency Room. Emerg Med Clin North Am. 2002; 20 (1): 177 – 98. PMID: 11826633
For More on This Topic Checkout:
- Sarah Melendez at ALiEM: Bleeding and Hemophilia in the Pediatric ED
- Sean Fox at Pediatric EM Morsels: Hemophilia in the ED
- Andrew Sloas at PEM ED Podcast: Hemophilia Emergencies
Post Peer Reviewed By: Anand Swaminathan (Twitter: @EMSwami)
The post Hemophilia: What’s so Bloody Funny? appeared first on REBEL EM - Emergency Medicine Blog.